Edit detail for FAQ revision 4 of 4
| 1 2 3 4 | ||
|
Editor: damberger
Time: 2017/04/28 20:53:30 GMT+2 |
||
| Note: | ||
added:
"Tutorials index":Tutorials
FAQ for CARA
Frequently asked questions and their answers. Questions about the use of CARA should be put to Fred Damberger or the CDT. Bugs and Feature requests should be submitted to the CARA Forum.
Please also check KnownBugs and CARAcrashes.
NEXT This is the last topic in the complete tour of the Wiki.
Return to FrontPage
Questions:
Section I: Basics
- What is difference between Move Spin and Move Spin Alias ? What's the deal with all this Alias stuff ?
- I'm confused with all these Scopes and Windows. What are they doing ?
- Should I pick a Peak, a Spin or a System ?
- I have XEASY atom, peak and sequence lists. What do I do ?
- What is a spin link .. or .. how do I represent NOEs between spins ?
- Where can I find a list of command line shortcuts like "cp" ?
- How do I write out my chemical shifts ?
- How do I measure the line width of a peak ?
Section II: Advanced
- How can I make a preliminary assignment ?
- How do I make stereospecific assignments ?
- How can I set the folding/aliasing properties of a peak ?
- What is peak inference?
- What are ave, dev, and SDmult?
Section III: Bugs, Crashes, Problems
- CARA crashes/I found a bug/I want a feature! What can I do ?.
- CARA crashes when reading a spectrum
- CARA does not accept external calibrations
- When overwriting a spectrum with an external program, CARA may crash
- Show folded doesn't do anything.
- I cannot rotate an imported peaklist.
- I have a problem to assign labels to spins during backbone assignment with new spectra types (e.g. HNCO and HN(CA)CO ). The labels C and C-1 are not available in SynchroScope, StripScope etc.
- I do not see any data when I open my 3D HNHA or 3D 15NH-HSQC-NOESY with Systemscope. I get the message "out of spectrum". I can open both spectra with monoscope.
- Why can't I show orthogonal in SystemScope for my CCH-TOCSY spectrum. It works fine with my 3D 15N-resolved NOESY.
- In want to use nmrPipe spectra in CARA.
- When I assign or pick spins with i-1 labels like "CA-1" in Triple Resonance spectra, they dissapear.
- I get the error message "Cannot import: There are 4 ambiguous shifts for the following atom numbers (see message log) 217 4513 6713 8817" when I try use the menu item "Peaks-Import Alias Shifts" in MonoScope.
- I get the error message "Error in Matrix Calculation: An exception has been thrown Runtime error:- detected by Newmat: matrix is singular" when I try to apply Peaks > Integrate All
- What is the purpose of Peak Color and Color Codes for SpinLinks? Why don't peaks or SpinLinks display in the color I selected?
Section IV: Miscellaneous
Answers:
How can I make a preliminary assignment ?
Use the prefix ? before any label, e.g. ?HA instead of HA. This will indicate to CARA that the assignment is not final. Furthermore, the standard checks for consistency will be switched off for this atom, allowing you to assign several chemical shifts with ?HA. A final assignment of HA is only possible to one atom. pascal
How can I set the folding/aliasing properties of a peak ?
Unlike XEASY, CARA works with the real chemical shifts from the beginning. The trick is to pick the spin at its real chemical shift. See this page for more details. pascal
What is the difference between "Move Spin" and "Move Spin Alias" ? What's the deal with all this Alias stuff ?
In an ideal case, every spin has exactly the same resonance frequency (or chemical shift) in every spectrum. However due to different experimental or sample conditions, the chemical shift for a given spin may vary between spectra. The "real" chemical shift of a spin is defined the first time it is assigned in a spectrum (say spectrum #1). An Alias can be created for this spin in a spectrum where the shift deviates from the "real value" using "Move Spin Alias" instead of Move Spin. Any other such functions are logical extensions of this concept.
You should use aliases only if really necessary (not to microadjust every peak). This is because when you create an alias in a spectrum the position of peaks involving that spin in the spectrum become independent of the position of the related peak in other spectra. This makes it difficult to detect inconsistencies in your assignments.
The state of your aliases can be checked in the Repository Window->Projects->MyProject?->Spins. Any Spin with a node next to its name has aliases. You can inspect their values in this list by clicking on the node. In spectra, normally peaks are represented by a "+" symbol. Peaks constructed from spins with an alias (in any dimension) are represented with an "x". To remove the alias, select the peak and execute "move alias" without moving the cursor.
Aliases are important for import and export from/to XEASY and for structure calculation software. Instead of importing a chemical shift or peak list you can import an Alias Shift List, that is only valid in the context of a particular spectrum. This is done by executing import alias shifts for the spectrum in question opened with MonoScope. To export the aliases for this spectrum, right-click the spectrum in Projects->MyProject->Spectrum and select Export Atom List.. pascal
I'm confused about all these Scopes and Windows. What are they doing ?
This page contains a summary of the tools and their functions. pascal
Should I pick a Peak, a Spin or a System ?
It is important to note that usually it is clear from the context what kind of object you should pick (meaning: it's less confusing than in sounds ...).
A Peak is CARA's representation of a crosspeak. It is fully compatible with XEASY's concept of peaks. It can be associated with a Volume. A peak is always bound to a given spectrum. Peaks don't use most of CARA's advanced functionality, but are useful for integration and interaction with external programs like XEASY or ATNOS/CANDID. Unlike in XEASY, in CARA peaks are generated if at all at the end of a project for integration purposes.
A Spin represents the resonance frequency of a single atom. CARA can link Spins together and show expected peaks in a spectrum even if they were not picked manually, based on the spin system and residue type definitions. Such a dynamically generated peak is "inferred" from the database.
A System is a collection of Spins. If you pick a new System, several Spins are put into the new System (picking a System in an 15N-HSQC yields HN and N Spins). A System can be linked to neighboring systems. pascal
What is a spin link .. or .. how do I represent NOEs between spins ?
A spin link is CARAs way of representing NOEs or through-space connection between spins. Each spin link contains two entries listing the spin numbers of the two linked spins. Once a spin link is created, a NOE crosspeak will appear in any NOESY spectra displayed with HomoScope, PolyScope, StripScope, and SystemScope where it is expected between the linked spins . Spin links are created automatically in a NOESY spectrum when propose peak is used in the above scopes. If you have a peaklist from an XEASY project, you can also generate spin links for all the proton pairs appearing in each line of the peaklist. See the PolyScope Tutorials for details. Fred
Where can I find a list of command line shortcuts like "cp" ?.
Alexey Neumoin has provided a PDF file which lists shortcuts. You can also do the following: In any Scope, type "?" at the command line and hit return. Then click on "Message Log". You will see all shortcuts for the Scope in the lower right window of the explorer.
Fred
How do I write out my chemical shifts ?.
The easiest way is to right-click on the project in Cara-explorer and select "Export-Atom List". This will write out the chemical shifts as a "proton list" in xeasy format. If you are doing structure calculations, you will need to write out in a format compatible with the residue library of the program in question. In this case the CALUA script "WriteAssignments.lua" is appropriate. You can select between BMRB, DYANA and CYANA2 formats. This script is also acceptable for writing out a BMRB deposit. If you are analysing the chemical shifts, you may want to write them out with each atom type in a separate column (e.g. all "CA" in one column). In this case, try the CALUA script "WriteShiftsInColumns.lua". A similar script is available for writing out shifts for systems as input to the assignment program PACES. "ExportToPaces.lua". If you are making plots of deviations from random coil shifts then the scripts "ChemShiftDeviationsFile.lua" and "ChemShiftDeviationsPlot.lua" are useful. There are additional scripts available for other applications.
Fred
How do I measure the line width of a peak ?.
The easiest way is to shift-click at the left edge of the peak in the slice window and drag the mouse to the right edge. The width is displayed on the status line. w 0.041 (20.45) means the width is 0.041 ppm or 20.45 Hz.
Fred
CARA crashes/I found a bug/I want a feature! What can I do ?
Check this page for frequent bugs and workarounds. A separate page deals with some frequent causes of crashes.
Serious bugs should be immediately reported via bug tracker. Please include your email address, as much details as possible and attach all pertinent files ( e.g. param files, peaklists, repositories ...).
Why the name CARA?
There is a tradition of programs with womens names in the Wuthrich group (like DIANA). CARA stands for "Computer Aided Resonance Assignment" Don't confuse it with the other CARAs... See the CARAsynonyms.
CARA crashes when reading a spectrum
Look here for some hints and suggestions.
Rochus: Instead of crashing CARA now gives you an error message when loading the spectrum. Only valid spectra can be loaded.
CARA does not accept external calibrations
If you change a .param file using a text editor or XEASY while CARA is running, CARA will not be aware of this change, since it stores the calibration upon loading. You will need to replace the spectrum with itself. Click on the Spectrum in the Spectra-Explorer. Right-click Replace Spectrum and reselect the spectrum to load it again.
When overwriting a spectrum with an external program, CARA may crash
A typical situation: You have opened a spectrum with Cara. Then you retransform the spectrum with PROSA and overwrite it. Then you open MonoScope. This may lead to a crash. Since CARA uses a "map" of the spectrum for fast navigation, retransforming and overwriting the spectrum may change this map. The solution is to replace the spectrum with itself. Click on the Spectrum in the Spectra-Explorer. Right-click Replace Spectrum and reselect the spectrum to load it again.
Show folded doesn't do anything.
Your .param file is bad. This may happen with .param files generated with uxnmr2xeasy.
Set Folding in w1 ................. NO to the correct value, e.g. Folding in w1 ................. RSH
I cannot rotate an imported peaklist.
After importing a peaklist, you can rotate it. However as soon as you save it into the repository using Save Peaklist..., CARA considers the orientation to be final. So, the trick is to import, rotate and then save.
I have XEASY atom, peak and sequence lists. What do I do ?
See here . Don't forget to read the Tutorials to be able to use CARA's new and advanced features.
I have a problem to assign labels to spins during backbone assignment with new spectra types (e.g. HNCO and HN(CA)CO ). The labels C and C-1 are not available in SynchroScope, StripScope etc..
This is probably a spectrum type definition problem. See CreateSpectrumType for a general introduction to setting up spectrum types. Specifically for the HNCO & HN(CA)CO, try the following steps:
- In the Spectrum Types pane, Execute Add new type. Name it HN(CA)CO
- Set X->H, Y->C, Z->N. You can give names to the dimensions like HN, CO, N.
- Press Ok
- In case of a backbone assignment experiment, you have to work with labels. So click on each of the dimensions: Execute Add Label... and add HN for the Dim D1, C, C-1 for Dim D2 and N for Dim D3. (You can compare to the HNCA.) For the HNCO define only C-1 in Dim D2.
- For the scopes using Peak inference (HomoScope, PolyScope, StripScope, SystemScope) you will need to define a generic residue in the ResidueType pane of the CARA explorer. Right-click on a ResidueType which contains all the backbone atoms (e.g. LYS) and select generic residue. Second right-click again and select define termini. Enter N for N and C for C. This will allow CARAs peak inference engine to find atoms in sequentially neighboring residues (such as C-1).
How do I make stereo-specific assignments ?
When you make an assignment like "HB2" in CARA this means that you are assigning one of two HB atoms but you do not know whether it is HB2 or HB3. If you assign "QB" (Start1.1.cara)/ "HB" (Start1.2.cara) (group = HB2,HB3) this means that both HB2 and HB3 have identical chemical shifts. To indicate that an HB2 is stereoassigned, you place an exclamation point in front of the label: "!HB2".
Fred
What is peak inference?
Peak inference is a set of algorithms CARA uses to predict the positions of peaks in displayed spectra. It is (currently) active in the following scopes: HomoScope, PolyScope, StripScope, SystemScope. CARA turns the usual paradigm for resonance assignment on its head. Usually (e.g. XEASY) one picks peaks and by comparison of the correlations between different peaks they are eventually assigned a label and finally a residue. CARA from the beginning works with spins which each have a specific ppm value. Peaks are inferred from the spinlist and a knowledge-base which represents the magnetization-transfer pathway for each experiment, the SpectrumTypes?, and an atomic model for each ResidueType. CARA predicts the expected positions of the peaks when the spectrum is displayed. In order for all peaks to be correctly modeled, the label and ResidueType assigned to each spin must be correct. CARA thus continually monitors the consistency of the emerging database of spins and represents this in the form of inferred peaks in the displayed spectra. This scheme also avoids the need to create peaklists for each spectrum and the inevitable problems related to synchronizing these lists. Fred
What are ave, dev, and SDmult?
ave is the statistically-averaged chemical shift for a given atom from a ResidueType (usually the BMRB database value). dev is the range of deviations from ave which are considered close enough so that the shift of a given spin "matches" the statistically expected range for the atom that it is assigned to. The closer the shift is to ave, the higher the fitness of the match. Spins outside of dev, make zero fitness contribution in a fragment alignment. SDmult is the multiplier used by the script "LoadBmrbStats.lua" to calculate dev from the standard deviation of the BMRB database. E.g. the default value for SDmult = 4 meaning that dev = 4 SD where SD is the BMRB standard deviation for the chemical shifts assigned to the corresponding Atom in a given ResidueType. When "show alignment" is executed in StripScope, the selected fragment is mapped onto all possible sequence positions and for each position the fitness of every spin whose label matches an atom at the aligned residue position contributes to the total fitness for the fragment mapped at that position. This total fitness determines the ranking of the aligned fragment positions. Fred*
I do not see any data when I open my 3D HNHA or 3D 15NH-HSQC-NOESY with Systemscope. I get the message "out of spectrum". I can open both spectra with monoscope.
Do you mean that SystemScope does not show you any contours in the spectrum windows? Do you see a list of spins when you select a residue from the top menu? Do links between these spins appear in the bottom window? Have you checked the orientation of the spectra in MonoScope? The 3D HNHA should display x=HN dim, y=HA dim, z=N dim. If this is not the case, try using the Menu Spectrum-Map to Type to adjust the orientation of the spectrum with respect to the SpectrumType model. For the 3D 15NH-HSQC-NOESY the orientation should be x=HN dim, y=Hnoe dim, z=N dim. Check your SpectrumType definitions for the two spectra against the newest CARA template from the download page. Possibly there is an error in your SpectrumType definition. Fred
Why can't I "show orthogonal" in SystemScope for my CCH-TOCSY spectrum. It works fine with my 3D 15N-resolved NOESY.
To use the CCH-TOCSY in SystemScope as you described, open SystemScope(rotated) and switch the Cinept dimension (D3) to be along Dim X (horizontal), and the Hinept dimension (D1) to be along Dim Z (depth).
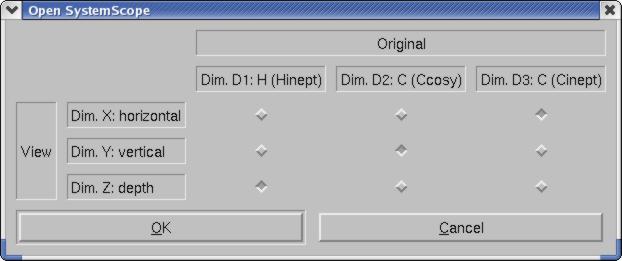
The reason it did not work for you in the standard orientation is the following:
Show Orthogonal displays a plane along Dim Y and Dim Z at the Dim Y position defined by the vertical position of the cursor in the strip.
This means that to use Show Orthogonal, the horizontal and vertical axes of the strip must have the same AtomType. Otherwise the position of the orthogonal plane is not defined. Fred.
I want to use nmrPipe spectra in CARA.
Starting with Cara 1.2 nmrPipe format spectra (*.pipe) can be read by CARA.
3D and 4D spectra: Use the program xyz2pipe supplied with the nmrPipe distribution to convert the individual plane files from nmrPipe to a single file.
E.g. for a 3D: xyz2pipe -in hnco%03d.ft -x > hnco_3D.ft
mv hnco_3D.ft hnco_3D.pipe
Fred
When I assign or pick spins with i-1 labels like "CA-1" in Triple Resonance spectra, they dissapear.
This is a problem which occurs with obsolete templates (like template_pascal_0.9.9.cara) which do not have parameters defined for inferring peaks of sequentially neighboring residues. New projects should not be started with old(obsolete) templates to avoid problems.
To fix the problem:
- Click on "ResidueTypes?" in the repository window of Cara Explorer. The residue types appear in the right window (Components Window).
- Right-Click in the Components Window and select "Set Terminals"
- Enter "N" for N-terminal i-1 and "C" for C-terminal i+1. This tells Cara how the Residues are connected to eachother in the sequence.
- Right-Click on a residue (like LYS) and select Generic Residue. This tells Cara what ResidueType to use as a default (when a spin-system is not yet assigned to a ResidueType) to do Peak Inference. I use LYS since it is a long residue which has all the typical labels.
- You may also have a problem if the SpectrumTypes? are defined in the wrong order. ExperimentProcedure? should always start on the own residue and only spread into other residues at a later stage. Example HNCA
a. Click on SpectrumType in the repository window of Cara Explorer. The SpectrumTypes? appear in the Components window.
b. Right-Click on "HNCA" and select Edit Procedure.
c. Check that the experiment has the order Step1(HN)->Step2(N)->Step3(Ca).
d. If the order is reversed Step1(Ca)->Step2(N)->Step3(HN), you can fix this as follows: Right-Click on "HNCA" in the SpectrumTypes? components window of Cara-Explorer and select "Reverse Steps"
After executing steps a-c and if necessary step d, your i-1 spins should appear in freshly opened scopes. The scopes opened BEFORE you made these changes will not register the change and so don't show the i-1 spins. Close these old scopes. Fred
I get the error message "Cannot import: There are 4 ambiguous shifts for the following atom numbers (see message log) 217 4513 6713 8817" when I try use the menu item "Peaks-Import Alias Shifts" in MonoScope.
This occurs because CARA expects all atoms to have one and only one chemical shift. Your peaklist has different shifts for the same atom numbers. E.g. if it is a 3D NOESY peaklist, it might be that the shift for atom 217 ("HN of residue 10") has a different shift 8.702 in the direct dimension for a given peak, than it has for another peak in the indirect dimension 8.709.
There are two workarounds for this:
- You can change the peaklist so that every occurance of a given atom has the same exact shift. Then CARA should generate the aliases using "Peaks-Import Alias Shifts" without problems.
- You can use a LUA script I wrote to import the aliases. The script simply ignores the inconsistencies and uses the last occurance of the atom in the peaklist to define the chemical shift. If you know that the differences are small/insignificant this is the fastest way to solve the problem. Note that a number of modifications of the script could make it more sophisticated. It could for example take an average shift from all occurances of a given atom, or it could use only the direct dimension. Etc. These modifications I leave as an exercise to the user.
Fred
I get the error message "Error in Matrix Calculation" when I try to Integrate all in MonoScope.
This error message means that at least two peaks in the peaklist are at identical positions. The matrix calculation CARA uses to determine the intensities belonging to individual signals fails in this case - and the Newmat library returns an error. The quick way to solve this problem is to use the latest version of Cara (version 1.3 and up). These newer versions prescreen the peak positions and divide intensity up equally among the degenerate peaks. To remove degenerate peaks (or move one slightly so they are nolonger degenerate) use the menu item Peaks > Check for Doubles. This will write the peak numbers for pairs of degenerate peaks to the Message Log window of Cara-Explorer. Click on the Message Log window in Cara-explorer and write down the pairs. Now double-click on the peak number in the peaklist of MonoScope to select one peak. You can now either (a) delete the peak "dp" or (b) move the cursor with arrow keys and then "mp" to move the peak slightly. After doing this for one peak in each degenerate pair. Repeat the Peaks > Integrate All.
Fred
Each SpinLink has a "code" that can be set to an integer value. You set the value by right-clicking the o-o symbol and selecting "Set link params". This affects the color of the displayed SpinLink symbol "o-o". All SpinLinks with the same integer value for the "code" parameter will display in the same color.
You determine the color code (i.e. which integer corresponds to which color) in MonoScope by right-clicking on a peak in the PeakList? window and "Select color code".
In CARA version 1.8.4 there are some display problems with color codes. Peaks and SpinLink symbols are displayed with the default color instead of the selected color. In addition, the SpinLink? symbol dissapears if the color is greater than 6. This is fixed in cara 1.9.0 beta.
The purpose of the "color" parameter is to identify different catagories of peaks (or SpinLinks). Programs such as XEASY, ATNOS/CANDID and CYANA/NOEASSIGN generate peaklists where each peak can have a color (values from 1 to 6 are allowed). These color are often used to indicate peaks with different properties. E.g. low assignment reliability, unassigned, or similar.
The color of the peak has no affect on the peaklist that is written out by CARA except that the color values of the peaks in the peaklist that is written out are equal to the values you set within CARA. In XEASY (or NEASY) the color value can be used to change the display color of all peaks with a given color value. ATNOS/CANDID and CYANA/NOEASSIGN do not care what the peak color value is.