Edit detail for SpectrumTypeCBHD revision 1 of 1
| 1 | ||
|
Editor: damberger
Time: 2007/01/22 12:41:05 GMT+0 |
||
| Note: links updated after rename | ||
changed: - A complex example of SpectrumType (CBHD Yamazaki Experiment): As a more complex example we describe step-by-step how to set up a SpectrumType for the CBHD experiment correlating CB and HD of aromatic residues. Create the spectrum type Use *Add Type...* to enter a new type or *Duplicate Type...* and then *Edit Type...* to base your new type on an existing similar type. (It must use the same Atom Types along the same axes)<br> <img src="newtype1.gif" /> <br> Enter the Dimensions of the experiment in the orientation CARA should use. D1 is along X, D3 is depth, D2 is along Y (strip axis). NOTE: The screen shots were obtained when CARA still number the dimensions starting with Zero "0". For Additional examples: See the defined Spectrum Types in the template CARA files available on the TemplatesPage. Edit Procedure Next select the new spectrum type and use *Edit Procedure* to define how magnetization is transferred between spins in the experiment. <img src="newtype2.gif" /><br> Create all steps you want to include in your definition. This can include several editing dimensions. In many cases, you can reproduce the layout of your actual NMR experiment with one caveat: You must start the experiment in the System of interest. Branching to other systems must occur in later steps. E.g. HNCA should start on H(Step0)->N(Step1)->C(Step2) because in Step2 the magnetization can go to the predecessor system (CA-1). The experiment begins with Step 0. It will connect the atom type of step 0 with the atom type of step 1 by no more than the number of bonds as defined by *Hops*. Note: The number of *hops* for step 0 is irrelevant, since this is the starting step. *Hops*=-1 indicates a NOESY-type transfer. The Repeat-Option allows TOCSY-style experiments: E.g. a standard 2D-![H,H]-TOCSY is produced with the following definition: Step 0: *Target* 0=H Step 1: *Target* 1=H, *Hops*=3, and *Repeat*=On . *Mean* and *Deviation* are used to simulate selective excitations. *Mean* gives the center of the band excited in the step and *Deviation* gives the maximum offset from the center of the band which is excited during the step. Both values are given in ppm. This can be used to exclude 13C-aromatic resonances in a 3D 13C-aliphatic-resolved ![H,H]-NOESY, or for more complex situations as in the shown example. *Dimension* is used to map a given step onto a dimension of the spectrum. An empty Dimension-field denotes an editing step. *Comment* can be used to provide a name for this dimension. It is recommended to use the same name as in the Type Definition. The dimension numbering in a 3D should be as follows: D1 for the direct proton dimension, D3 for the attached heavy atom, D2 for the dimension in y-direction. Usually D1 and D3 define your anchor (HN,N or HC,C) and D2 the depth. If your spectrum appears wrongly oriented or the calculated anchors are not what you expected (CB,HD instead of CB,HB in the second example below), you probably mixed up the dimensions. Check your SpectrumType definition: *Show Experiment Path...* runs a check of your definition. Select a nonaromatic residue, like ALA, will give the following result for the CBHD as defined above. <b>Each step in the definition produces further indent</b>. <img src="newtype3.gif" /><br> This shows only diagonal peaks in the simulation. Diagonal peaks are always shown for technical reasons. Selecting the aromatic residue like Trp will yield: <img src="newtype4.gif" /><br> Note that a path from QB to HD1 is now possible. Any other non-diagonal combination is suppressed.
A complex example of SpectrumType (CBHD Yamazaki Experiment):
As a more complex example we describe step-by-step how to set up a SpectrumType for the CBHD experiment correlating CB and HD of aromatic residues.
Create the spectrum type
Use Add Type... to enter a new type or Duplicate Type... and then Edit Type... to base your new type on an existing similar type. (It must use the same Atom Types along the same axes)
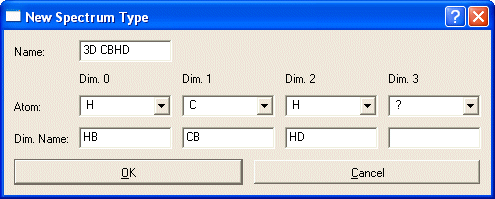
Enter the Dimensions of the experiment in the orientation CARA should use. D1 is along X, D3 is depth, D2 is along Y (strip axis). NOTE: The screen shots were obtained when CARA still number the dimensions starting with Zero "0".
For Additional examples: See the defined Spectrum Types in the template CARA files available on the TemplatesPage.
Edit Procedure
Next select the new spectrum type and use Edit Procedure to define how magnetization is transferred between spins in the experiment.
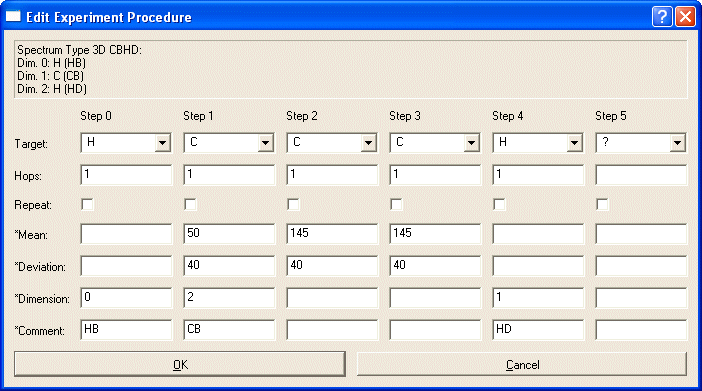
Create all steps you want to include in your definition. This can include several editing dimensions. In many cases, you can reproduce the layout of your actual NMR experiment with one caveat: You must start the experiment in the System of interest. Branching to other systems must occur in later steps. E.g. HNCA should start on H(Step0)->N(Step1)->C(Step2) because in Step2 the magnetization can go to the predecessor system (CA-1).
The experiment begins with Step 0. It will connect the atom type of step 0 with the atom type of step 1 by no more than the number of bonds as defined by Hops. Note: The number of hops for step 0 is irrelevant, since this is the starting step. Hops=-1 indicates a NOESY-type transfer. The Repeat-Option allows TOCSY-style experiments:
E.g. a standard 2D-[H,H]-TOCSY is produced with the following definition:
Step 0: Target 0=H
Step 1: Target 1=H, Hops=3, and Repeat=On .
Mean and Deviation are used to simulate selective excitations. Mean gives the center of the band excited in the step and Deviation gives the maximum offset from the center of the band which is excited during the step. Both values are given in ppm. This can be used to exclude 13C-aromatic resonances in a 3D 13C-aliphatic-resolved [H,H]-NOESY, or for more complex situations as in the shown example.
Dimension is used to map a given step onto a dimension of the spectrum. An empty Dimension-field denotes an editing step. Comment can be used to provide a name for this dimension. It is recommended to use the same name as in the Type Definition. The dimension numbering in a 3D should be as follows: D1 for the direct proton dimension, D3 for the attached heavy atom, D2 for the dimension in y-direction. Usually D1 and D3 define your anchor (HN,N or HC,C) and D2 the depth. If your spectrum appears wrongly oriented or the calculated anchors are not what you expected (CB,HD instead of CB,HB in the second example below), you probably mixed up the dimensions.
Check your SpectrumType definition:
Show Experiment Path... runs a check of your definition. Select a nonaromatic residue, like ALA, will give the following result for the CBHD as defined above. Each step in the definition produces further indent.
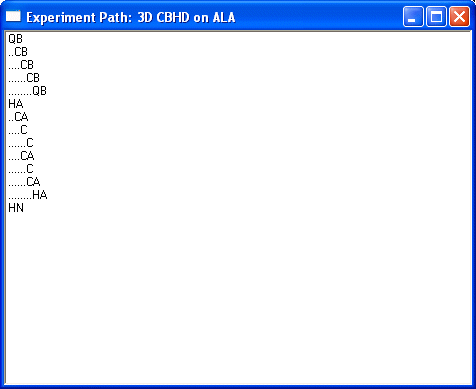
This shows only diagonal peaks in the simulation. Diagonal peaks are always shown for technical reasons. Selecting the aromatic residue like Trp will yield:
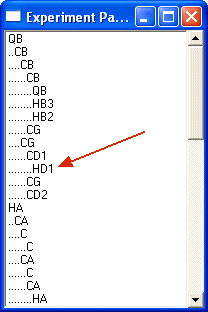
Note that a path from QB to HD1 is now possible. Any other non-diagonal combination is suppressed.